
AWS HealthOmics로 Nf-core 워크플로우 마이그레이션 하기 (rnaseq)
참고문서: https://catalog.us-east-1.prod.workshops.aws/workshops/76d4a4ff-fe6f-436a-a1c2-f7ce44bc5d17/en-US
이 문서를 참고하여 환경을 준비합니다. 여기서는 이 과정은 생략합니다.
프로젝트 셋업
nf-core repository로부터 워크플로우 복제
cd ~
git clone https://github.com/nf-core/rnaseq --branch 3.14.0 --single-branchDocker Image Manifest의 생성
cp ~/amazon-ecr-helper-for-aws-healthomics/lib/lambda/parse-image-uri/public_registry_properties.json namespace.configinspect_nf.py 를 실행합니다.
python3 amazon-omics-tutorials/utils/scripts/inspect_nf.py \
--output-manifest-file rnaseq_3140_docker_images_manifest.json \
-n namespace.config \
--output-config-file omics.config \
--region <region> \
~/rnaseq/
생성되는 두 개의 출력은 rnaseq_3140_docker_images_manifest.json 과 omics.config입니다.
rnaseq_3140_docker_images_manifest.json 파일은 다음과 같은 모습이어야 합니다:
{
"manifest": [
"quay.io/biocontainers/bbmap:39.01--h5c4e2a8_0",
"quay.io/biocontainers/bedtools:2.30.0--hc088bd4_0",
"quay.io/biocontainers/bioconductor-dupradar:1.28.0--r42hdfd78af_0",
"quay.io/biocontainers/bioconductor-summarizedexperiment:1.24.0--r41hdfd78af_0",
"quay.io/biocontainers/bioconductor-tximeta:1.12.0--r41hdfd78af_0",
"quay.io/biocontainers/fastp:0.23.4--h5f740d0_0",
"quay.io/biocontainers/fastqc:0.12.1--hdfd78af_0",
"quay.io/biocontainers/fq:0.9.1--h9ee0642_0",
"quay.io/biocontainers/gffread:0.12.1--h8b12597_0",
"quay.io/biocontainers/hisat2:2.2.1--h1b792b2_3",
"quay.io/biocontainers/kallisto:0.48.0--h15996b6_2",
"quay.io/biocontainers/mulled-v2-1fa26d1ce03c295fe2fdcf85831a92fbcbd7e8c2:1df389393721fc66f3fd8778ad938ac711951107-0",
"quay.io/biocontainers/mulled-v2-1fa26d1ce03c295fe2fdcf85831a92fbcbd7e8c2:59cdd445419f14abac76b31dd0d71217994cbcc9-0",
"quay.io/biocontainers/mulled-v2-8849acf39a43cdd6c839a369a74c0adc823e2f91:ab110436faf952a33575c64dd74615a84011450b-0",
"quay.io/biocontainers/mulled-v2-a97e90b3b802d1da3d6958e0867610c718cb5eb1:2cdf6bf1e92acbeb9b2834b1c58754167173a410-0",
"quay.io/biocontainers/mulled-v2-cf0123ef83b3c38c13e3b0696a3f285d3f20f15b:64aad4a4e144878400649e71f42105311be7ed87-0",
"quay.io/biocontainers/multiqc:1.19--pyhdfd78af_0",
"quay.io/biocontainers/perl:5.26.2",
"quay.io/biocontainers/picard:3.0.0--hdfd78af_1",
"quay.io/biocontainers/preseq:3.1.2--h445547b_2",
"quay.io/biocontainers/python:3.9--1",
"quay.io/biocontainers/qualimap:2.3--hdfd78af_0",
"quay.io/biocontainers/rseqc:5.0.3--py39hf95cd2a_0",
"quay.io/biocontainers/salmon:1.10.1--h7e5ed60_0",
"quay.io/biocontainers/samtools:1.16.1--h6899075_1",
"quay.io/biocontainers/samtools:1.17--h00cdaf9_0",
"quay.io/biocontainers/sortmerna:4.3.4--h9ee0642_0",
"quay.io/biocontainers/stringtie:2.2.1--hecb563c_2",
"quay.io/biocontainers/subread:2.0.1--hed695b0_0",
"quay.io/biocontainers/trim-galore:0.6.7--hdfd78af_0",
"quay.io/biocontainers/ucsc-bedclip:377--h0b8a92a_2",
"quay.io/biocontainers/ucsc-bedgraphtobigwig:445--h954228d_0",
"quay.io/biocontainers/umi_tools:1.1.4--py38hbff2b2d_1",
"quay.io/nf-core/ubuntu:20.04"
]
}컨테이너 사설화
aws stepfunctions start-execution \
--state-machine-arn arn:aws:states:<region>:<your-account-id>:stateMachine:omx-container-puller \
--input file://rnaseq_3140_docker_images_manifest.json
nf-core project 코드 업데이트
mv omics.config rnaseq/conf
echo "includeConfig 'conf/omics.config'" >> rnaseq/nextflow.config
AWS HealthOmics 워크플로우 만들기
단계1. AWS HealthOmics 파라미터 파일
paramter-descrption.json을 만들어 아래와 같이 저장합니다.
새 워크플로마다 다른 매개변수를 사용할 수 있습니다. 위의 매개변수 템플릿 파일은 SCRNA-Seq nf-core 워크플로에만 해당됩니다. 새 파라미터 파일을 만들 때는 이 워크플로우에 대한 파라미터를 확인해야 합니다.
NF-Core 워크플로우에 대한 파라미터를 찾으려면 설명서를 참조하세요(예: RNA-Seq 파이프라인 파라미터는 여기).
{
"input": {
"description": "S3 URI to samplesheet.csv. Rows therein point to S3 URIs for fastq data",
"optional": false
},
"genome": {
"description": "Name of iGenomes reference. - e.g. GRCh38",
"optional": true
},
"fasta": {
"description": "Path to FASTA genome file. This parameter is mandatory if --genome is not specified. If you don't have the appropriate alignment index available this will be generated for you automatically.",
"optional": true
},
"gtf": {
"description": "Path to GTF annotation file. This parameter is mandatory if --genome is not specified.",
"optional": true
},
"gff": {
"description": "Path to GFF3 annotation file. This parameter must be specified if `--genome` or `--gtf` are not specified.",
"optional": true
},
"pseudo_aligner": {
"description": "Specifies the pseudo aligner to use - available options are 'salmon'. Runs in addition to `--aligner`",
"optional": true
}
}단계2. 워크플로우 스테이징
zip -r rnaseq-workflow.zip rnaseqaws s3 cp rnaseq-workflow.zip s3://<yourbucket>/workshop/rnaseq-workflow.zip
aws omics create-workflow \
--name rnaseq-v3140 \
--definition-uri s3://<yourbucket>//workshop/rnaseq-workflow.zip \
--parameter-template file://parameter-description.json \
--engine NEXTFLOW단계3. 워크플로우 생성 확인
aws omics list-workflows --name rnaseq-v3140워크플로우 테스트하기
입력파일 준비
아래의 입력 파일들은 여기를 참고하여 준비할 수 있습니다.
wget https://raw.githubusercontent.com/nf-core/test-datasets/rnaseq/samplesheet/v3.10/samplesheet_full.csv
aws s3 mv samplesheet_full.csv s3://{mybucket}/workshop/parameter-description.json에 사용된 것과 동일한 키를 사용하여 input_rnaseq.json 파일을 새로 만듭니다. 값은 워크플로에서 허용되는 실제 S3 경로 또는 문자열이 됩니다.
{
"input": "s3://{mybucket}/workshop/samplesheet_full.csv",
"genome": "GRCh37",
"pseudo_aligner": "salmon"
}위의 입력데이터로 필요한 S3 버킷에 대해 아래 Policy 준비에 반영되어야 합니다.
마찬가지로 결과 데이터를 작성할 버킷에 대한 권한도 Policy 준비에 반영해주세요.
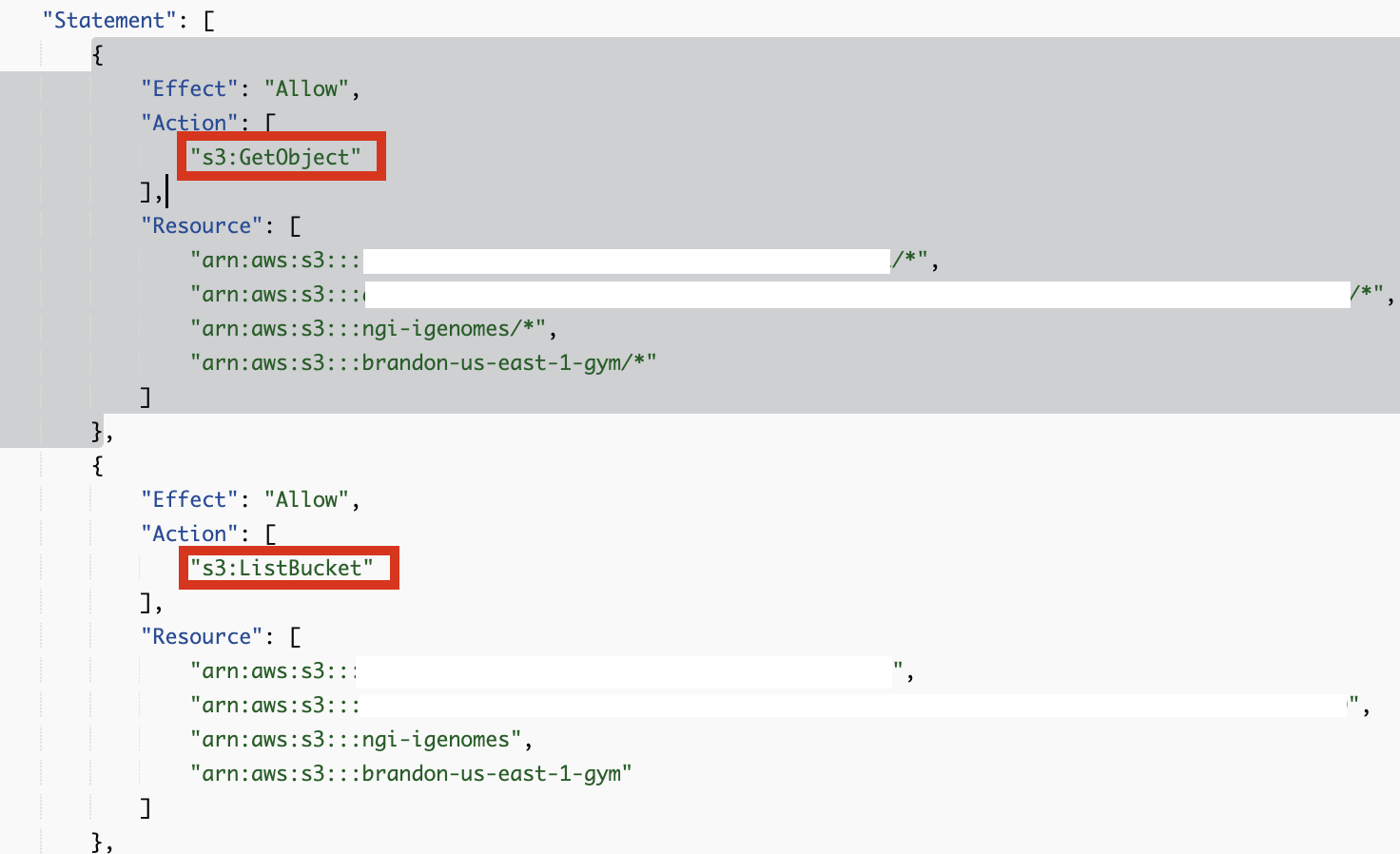
s3:GetObject, s3:ListBucket 권한
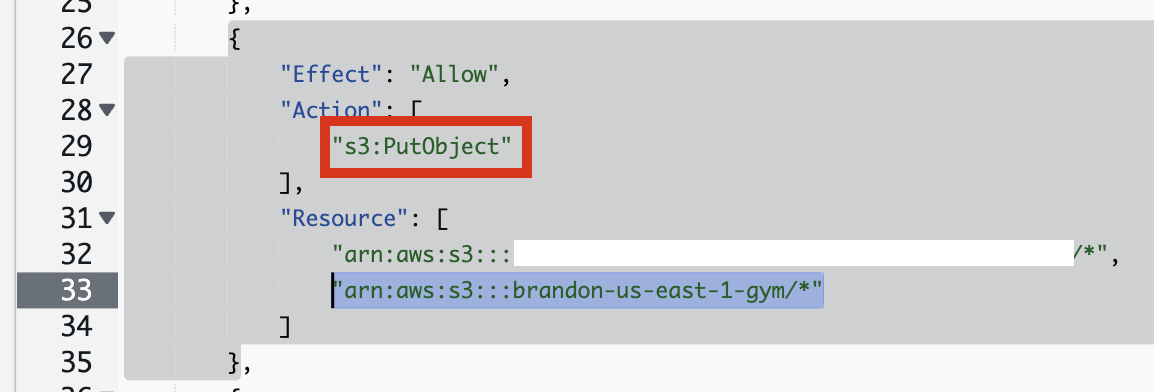
s3:PutObject 권한
Policy 준비
Prepare IAM service role to run AWS HealthOmics workflow
omics_workflow_policy.json 만들기
# 환경 변수 설정
export yourbucket="your-bucket-name"
export your_account_id="your-account-id"
export region="your-region"
# JSON 내용 생성 및 파일로 저장
cat << EOF > omics_workflow_policy.json
{
"Version": "2012-10-17",
"Statement": [
{
"Effect": "Allow",
"Action": [
"s3:GetObject"
],
"Resource": [
"arn:aws:s3:::${yourbucket}/*",
"arn:aws:s3:::${youranotherbucket}/workshop/nfcore-rnaseq-v3.14.0/*"
]
},
{
"Effect": "Allow",
"Action": [
"s3:ListBucket"
],
"Resource": [
"arn:aws:s3:::${yourbucket}",
"arn:aws:s3:::${youranotherbucket}/workshop/nfcore-rnaseq-v3.14.0"
]
},
{
"Effect": "Allow",
"Action": [
"s3:PutObject"
],
"Resource": [
"arn:aws:s3:::${yourbucket}/*"
]
},
{
"Effect": "Allow",
"Action": [
"logs:DescribeLogStreams",
"logs:CreateLogStream",
"logs:PutLogEvents"
],
"Resource": [
"arn:aws:logs:${region}:${your_account_id}:log-group:/aws/omics/WorkflowLog:log-stream:*"
]
},
{
"Effect": "Allow",
"Action": [
"logs:CreateLogGroup"
],
"Resource": [
"arn:aws:logs:${region}:${your_account_id}:log-group:/aws/omics/WorkflowLog:*"
]
},
{
"Effect": "Allow",
"Action": [
"ecr:BatchGetImage",
"ecr:GetDownloadUrlForLayer",
"ecr:BatchCheckLayerAvailability"
],
"Resource": [
"arn:aws:ecr:${region}:${your_account_id}:repository/*"
]
}
]
}
EOF
echo "omics_workflow_policy.json 파일이 생성되었습니다."trust_policy.json 만들기
# 환경 변수 설정
export your_account_id="your-account-id"
export region="us-east-1" # 기본값으로 us-east-1을 설정했습니다. 필요시 변경하세요.
# JSON 내용 생성 및 파일로 저장
cat << EOF > trust_policy.json
{
"Version": "2012-10-17",
"Statement": [
{
"Effect": "Allow",
"Principal": {
"Service": "omics.amazonaws.com"
},
"Action": "sts:AssumeRole",
"Condition": {
"StringEquals": {
"aws:SourceAccount": "${your_account_id}"
},
"ArnLike": {
"aws:SourceArn": "arn:aws:omics:${region}:${your_account_id}:run/*"
}
}
}
]
}
EOF
echo "trust_policy.json 파일이 생성되었습니다."IAM Role 생성
aws iam create-role --role-name omics_start_run_role_v1 --assume-role-policy-document file://trust_policy.json
Policy document 생성
aws iam put-role-policy --role-name omics_start_run_role_v1 --policy-name OmicsWorkflowV1 --policy-document file://omics_workflow_policy.json
워크플로우 실행
aws omics start-run \
--name rnaseq_workshop_test_run_1 \
--role-arn arn:aws:iam::<your-account-id>:role/omics_start_run_role_v1 \
--workflow-id <your-workflow-id> \
--parameters file://input_rnaseq.json \
--output-uri s3://<yourbucket>/output/